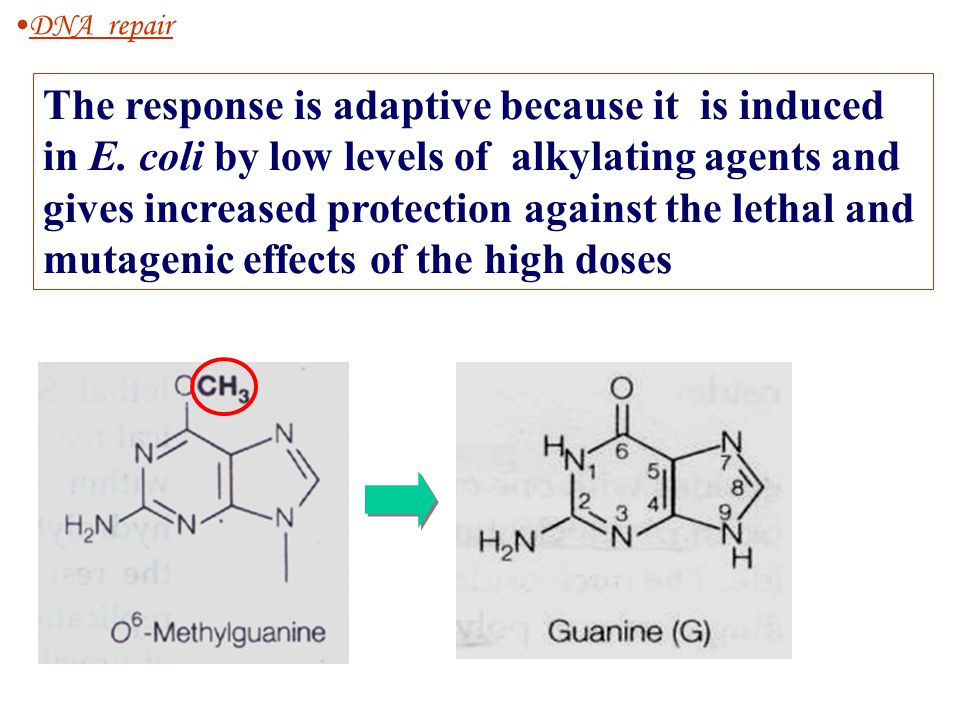
DNA repair
4 stars based on
52 reviews
DNA repair is a collection of processes by which a cell identifies and corrects damage to the DNA molecules that encode its genome. In human cells, both normal metabolic activities and environmental factors such as radiation can cause DNA damage, resulting in as many as 1 million individual molecular lesions per cell per day.
Other lesions induce potentially harmful mutations in the cell's genome, which affect the survival of its daughter cells after it undergoes mitosis. When normal repair processes fail, and when cellular apoptosis does not occur, irreparable DNA damage may occur, including double-strand breaks and DNA crosslinkages recombination repair and direct reversal crosslinks or ICLs.
The rate of DNA repair is dependent on many factors, including the cell type, the age of the cell, and the extracellular environment. A cell that has accumulated a large amount of DNA damage, or one that no longer effectively repairs damage incurred to its DNA, can enter one of three possible states:. The DNA repair ability of a cell is vital to the integrity of its genome and thus to the normal functionality of that organism.
Many genes that were initially shown to influence life span have turned out to be involved in DNA damage repair and protection. DNA damage, due to environmental factors and normal metabolic processes inside the cell, occurs at a rate of 10, to 1, molecular lesions per cell per day. The vast majority of DNA damage affects the primary structure of recombination repair and direct reversal double helix; that is, the bases themselves are chemically modified.
These modifications can in turn disrupt the molecules' regular helical structure by introducing non-native chemical bonds or bulky adducts that do not fit in the standard double helix.
Unlike proteins and RNADNA usually lacks tertiary structure and therefore damage or disturbance does not occur at that level. DNA is, however, supercoiled and wound around "packaging" proteins called histones in eukaryotesand both superstructures are vulnerable to the effects of DNA damage.
The replication of damaged DNA before cell division can lead to the incorporation of wrong bases opposite damaged ones. Daughter cells that inherit these wrong bases carry mutations from which the original DNA sequence is unrecoverable except in the rare case of a back mutationfor example, through gene conversion.
Spontaneous damage can include the loss of a base, deamination, sugar ring puckering and tautomeric shift. Nuclear DNA nDNA exists as chromatin during non-replicative stages of the cell cycle and is condensed into aggregate structures known as chromosomes during cell division. In either state the DNA is highly compacted recombination repair and direct reversal wound up around bead-like proteins called histones. Whenever a cell needs to express the genetic information encoded in its nDNA the required chromosomal region is unravelled, genes located therein are expressed, and then the region is condensed back to its resting conformation.
Mitochondrial DNA mtDNA is located inside mitochondria organellesexists in multiple copies, and is also tightly associated with a number of proteins to form a complex known as the nucleoid. Inside mitochondria, reactive oxygen species ROSor free radicalsbyproducts of the constant production of adenosine triphosphate ATP via oxidative phosphorylationcreate a highly oxidative environment that is known to damage mtDNA. A critical enzyme in counteracting the toxicity of these species is superoxide dismutasewhich is present in both the mitochondria and cytoplasm of eukaryotic cells.
Senescence, an irreversible process in which the cell no longer dividesis a protective response to the shortening of the chromosome ends. The telomeres are long regions of repetitive noncoding DNA that cap chromosomes and undergo partial recombination repair and direct reversal each time a cell undergoes division see Hayflick limit. Senescence in cells may serve as a functional alternative to apoptosis in cases where the physical presence of a cell for spatial reasons is required by the organism, [11] which serves as a "last resort" mechanism to prevent a cell with damaged DNA from replicating inappropriately in the absence of pro-growth cellular signaling.
Unregulated cell division can lead to the formation of a tumor see cancerwhich is potentially lethal to an organism. Therefore, the induction of senescence and apoptosis is considered to be part of a strategy of protection against cancer. DNA damage and mutation are fundamentally different. Damage results in physical abnormalities in the DNA, such as single- and double-strand breaks, 8-hydroxydeoxyguanosine residues, and polycyclic aromatic hydrocarbon adducts.
DNA damage can be recombination repair and direct reversal by enzymes, recombination repair and direct reversal thus can be correctly repaired if redundant information, such as the undamaged sequence in the complementary DNA strand or in a homologous chromosome, is available for copying.
If a cell retains DNA damage, transcription of a gene can be prevented, and thus translation into a protein will also be blocked. Replication may also be blocked or the cell may die. A mutation cannot be recognized by enzymes once the base change is present in both DNA strands, and thus a mutation cannot be repaired. At the cellular level, mutations can recombination repair and direct reversal alterations in protein function and regulation.
Mutations are replicated when the cell replicates. In a population of cells, mutant cells will increase or decrease in frequency according to the effects of the mutation on the ability of the cell to survive and reproduce.
Although distinctly different from each other, DNA damage and mutation are related because DNA damage often causes errors of DNA synthesis during replication or repair; these errors are a major source of mutation. Given these properties of DNA damage and mutation, it can be seen that DNA damage is a special problem in non-dividing or slowly-dividing cells, where unrepaired damage will tend to accumulate over time.
On the other hand, in rapidly-dividing cells, unrepaired DNA damage that does not kill the cell by blocking replication will tend to cause replication errors and thus mutation. The great majority of mutations that are not neutral in their effect are deleterious to a cell's survival.
Thus, in a population of cells composing a tissue with replicating cells, mutant cells will tend to be lost. However, infrequent mutations that provide a survival advantage will tend recombination repair and direct reversal clonally expand at the expense of neighboring cells in the tissue.
This advantage to the cell is disadvantageous to the whole organism, because such recombination repair and direct reversal cells can give rise to cancer. Thus, DNA damage in frequently dividing cells, because it gives rise to mutations, is a prominent cause of cancer. In contrast, DNA damage in infrequently-dividing cells is likely a prominent cause of aging.
Cells cannot function if DNA damage corrupts the integrity and accessibility of essential information in the genome but cells remain superficially functional when non-essential genes are missing or damaged. Depending on the type of damage inflicted on the DNA's double helical structure, a variety of repair strategies have evolved to restore lost recombination repair and direct reversal.
If possible, cells use the unmodified complementary strand of the DNA or the sister chromatid as a template to recover the original information. Without access to a template, cells use an error-prone recovery mechanism known as translesion synthesis as a last resort. Damage to DNA alters the spatial configuration of the helix, and such alterations can be detected by the cell.
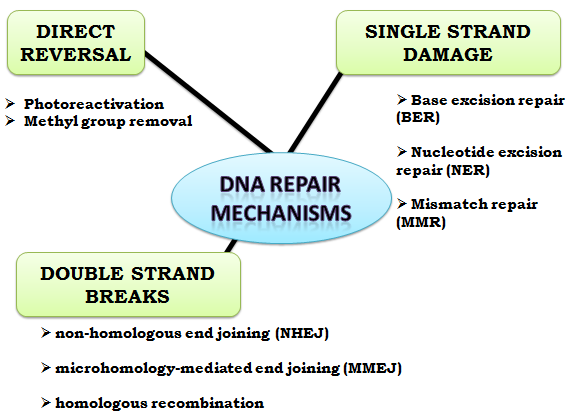
Once damage is localized, specific DNA repair molecules bind at or near the site of damage, inducing other molecules to bind and form a complex that enables the actual repair to take place. Cells are known to eliminate three types of damage to their DNA by chemically reversing it.
These mechanisms do not require recombination repair and direct reversal template, since the types of damage they counteract can occur in only one of the four bases. Such direct reversal mechanisms are specific to the type of damage incurred and do not involve breakage of the phosphodiester backbone. The formation of pyrimidine dimers upon irradiation with UV light results in an abnormal covalent bond between adjacent pyrimidine bases.
Another type of damage, methylation of guanine bases, is directly reversed by the protein methyl guanine methyl transferase MGMTthe bacterial equivalent of which is called ogt. This is an expensive process because each MGMT molecule can be used only once; that is, the reaction is stoichiometric rather than catalytic.
When only one of the two strands of a double helix has a defect, the other strand can be used as a template to guide the correction of the damaged strand. In order to repair damage to one of the two paired molecules of DNA, there exist a number of excision repair mechanisms that remove the damaged nucleotide and replace it with an undamaged nucleotide complementary to that found in the recombination repair and direct reversal DNA strand. Double-strand breaks, in which both strands in the double helix are severed, are particularly hazardous to the cell because they can lead to genome rearrangements.
PVN Acharya noted that double-strand breaks and a "cross-linkage joining both strands at the same point is irreparable because neither strand can then serve as a template for repair. The cell will die in the next mitosis or in some rare instances, mutate. If these overhangs are compatible, repair is usually accurate. Loss of damaged nucleotides at the break site can lead to deletions, and joining of nonmatching termini forms insertions or translocations. NHEJ is especially important before the cell has replicated its DNA, since there is no template available for repair by homologous recombination.
There are "backup" NHEJ pathways in higher eukaryotes. Homologous recombination requires the presence of an identical or nearly identical sequence to be used as a template for repair of the break. The enzymatic machinery responsible for this repair process is nearly identical to the machinery responsible for chromosomal crossover during meiosis. This pathway allows a damaged chromosome to be repaired using a sister chromatid available in G2 after DNA recombination repair and direct reversal or a homologous chromosome as a template.
DSBs caused by the replication machinery attempting to synthesize across a single-strand break or unrepaired lesion cause collapse of the replication fork and are typically repaired by recombination. MMEJ starts with short-range end resection by MRE11 nuclease on either side of a double-strand break to reveal microhomology regions. There is pairing of microhomology regions followed by recruitment of flap structure-specific endonuclease 1 FEN1 to remove overhanging flaps. The extremophile Deinococcus radiodurans has a remarkable ability to survive DNA damage from ionizing radiation and other sources.
Partially overlapping fragments are then used for synthesis of homologous regions through a moving D-loop that can continue extension until they find complementary partner strands. In the final step there is crossover by means of RecA -dependent homologous recombination. Topoisomerases introduce both single- and double-strand breaks in the course of changing the DNA's state of supercoilingwhich is especially common in regions near recombination repair and direct reversal open replication fork.
Such breaks are not considered DNA damage because they are a natural intermediate in the topoisomerase biochemical mechanism and are immediately repaired by the enzymes that created them. DNA polymerase IV or V, from the Y Polymerase familyoften with larger active sites that can facilitate the insertion of bases opposite damaged nucleotides.
The polymerase switching is thought to be mediated by, among other factors, the post-translational modification of recombination repair and direct reversal replication processivity factor PCNA. Translesion synthesis polymerases often have low fidelity high propensity to insert wrong bases on undamaged templates relative to regular polymerases. However, many are extremely efficient at inserting correct bases opposite specific types of damage. From a cellular perspective, risking the introduction of point mutations during translesion synthesis may be preferable to resorting to more drastic mechanisms of DNA repair, which may cause gross chromosomal aberrations or cell death.
Recombination repair and direct reversal short, the process involves specialized polymerases either bypassing or repairing lesions at locations of stalled DNA recombination repair and direct reversal. Cells exposed to ionizing radiationultraviolet light or chemicals are prone to acquire multiple sites of bulky DNA lesions and double-strand breaks.
The accumulation of damage, to be specific, double-strand breaks or adducts stalling the replication forksare among known stimulation signals for a global response to DNA damage. The common features of global response are induction of multiple genescell cycle arrest, and inhibition of cell division.
The packaging of eukaryotic DNA into chromatin presents a barrier to all DNA-based processes that require recruitment of enzymes to their sites of action. To allow DNA repair, the chromatin must be remodeled. In eukaryotes, ATP dependent chromatin remodeling complexes and histone-modifying enzymes are two predominant factors employed to accomplish this remodeling process.
Chromatin relaxation occurs rapidly at the site of a DNA damage. This relaxation allows other proteins in the nucleotide excision repair pathway to enter the chromatin and repair UV-induced cyclobutane pyrimidine dimer damages. After rapid chromatin remodelingcell cycle checkpoints are activated to allow DNA repair to occur before the cell cycle progresses.
This is followed by phosphorylation of the cell cycle checkpoint protein Chk1initiating its function, about 10 minutes after DNA is damaged. After DNA damage, cell cycle checkpoints are activated. Checkpoint activation recombination repair and direct reversal the cell cycle and gives the cell time to repair the damage before continuing to divide.
An intra- S checkpoint also exists. These kinases phosphorylate downstream targets in a signal transduction cascade, eventually leading to cell cycle arrest.
DNA damage checkpoint is a signal transduction pathway that blocks cell recombination repair and direct reversal progression in G1, G2 and metaphase and slows down the rate of S phase progression when DNA is damaged.
It leads to a pause in cell cycle allowing the cell time to repair the damage before continuing to divide.